Can I Respond to the Activation of Multi Roll
The adhesion of leukocytes to vascular endothelium is a hallmark of the inflammatory process. This recruitment process and the requirement for (and participation of) specific adhesion glycoproteins in the binding of leukocytes to ECs have been elegantly demonstrated using a variety of experimental approaches. Direct visualization of the inflamed microvasculature has revealed that as leukocytes exit capillaries, hemodynamic forces give rise to an outward radial movement of leukocytes toward the venular endothelium. This margination process is generally attributed to red blood cells (which normally pile up behind the larger leukocytes in capillaries) that overtake the leukocytes and tend to push them toward the venular wall. The initial adhesive interactions between the leukocytes and venular endothelium are tethering (capture) and rolling. These low-affinity (weak) interactions are subsequently strengthened as a result of leukocyte activation (mediated by chemokine-dependent and chemokine-independent mechanisms). Consequently, the leukocytes attach to the endothelium and remain stationary. The leukocytes are then able to migrate into the interstitium through spaces between adjacent ECs. These interactions are initiated by a variety of chemical mediators that are elaborated from inflamed tissue and the entire process of leukocyte–endothelial cell adhesion is regulated by the sequential activation of different families of adhesion molecules that are expressed on the surface of leukocytes and ECs. The current paradigm for leukocyte (neutrophil) recruitment in the inflamed microvasculature is summarized in Figure 7.1. The adhesive determinants are known to vary between vascular beds and between different leukocyte populations [17,37,46,120–123].

FIGURE 7.1
Multistep adhesion cascade and the major molecular contributors to leukocyte recruitment during an inflammatory response. Following endothelial cell activation and the increased expression of P- and E-selectins, low-affinity adhesive interactions (capture (more...)
7.1. Adhesion Molecules
Table 7.1 summarizes some of the adhesion molecules that are expressed on the surface of leukocytes and their respective counter-receptors on ECs. Lectin-like adhesion glycoproteins, called the selectins, mediate leukocyte rolling, while the firm adhesion and subsequent transendothelial migration of leukocytes are mediated by the interaction of integrins (CD11/CD18, VLA-4) on leukocytes with immunoglobulin-like adhesion molecules on ECs (e.g., ICAM-1, VCAM-1). The expression of P-selectin, E-selectin ICAM-1, and VCAM-l on venular EC are temporally coordinated to ensure that the processes of leukocyte rolling and firm adhesion/emigration can occur for several hours after the initiation of an inflammatory response. The importance of these endothelial cell adhesion molecules and their counter-receptors on leukocyte recruitment in different animal models of inflammation has been demonstrated using either adhesion molecule-specific blocking monoclonal antibodies (mAbs) or mice that are genetically deficient in one or more adhesion molecules [37,79,86,122–127].
TABLE 7.1
Leukocyte adhesion receptors and their ligands on activated ECs.
7.1.1. Intraorgan Heterogeneity of Adhesion
The sequential, coordinated recruitment of leukocytes into the inflamed microvasculature is largely confined to postcapillary venules, with leukocyte–endothelial cell adhesion rarely seen in arterioles. For example, in the rat mesenteric microcirculation, 39% of all leukocytes passing through venules are rolling, while only 0.6% of leukocytes roll in the upstream arterioles. The basis for the preferential binding of leukocytes to venular ECs during inflammation appears to relate to the higher expression of endothelial cell adhesion molecules (CAM) in venules. Immunohistochemical analyses of CAM expression in the microcirculation have yielded results that consistently show a preferential expression of endothelial CAMs in postcapillary venules. Endothelial cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), E-selectin, and P-selectin can be detected on the surface of activated ECs in arterioles and occasionally capillaries; however, the density of these adhesion molecules is far greater on venular endothelium. Quantitative information bearing on this issue has been generated using laser confocal microscopy to quantify the constitutive expression of ICAM-1 in arterioles, capillaries, and venules of rat mesentery, using an FITC-labeled anti-rat monoclonal antibody. Mesenteric venules exhibited a 10-fold higher density of ICAM-1 than in the upstream arterioles and capillaries. This approach also revealed heterogeneity of ICAM-1 expression within different sized venules. Venules with diameters of 25 µm appear to exhibit the greatest density of ICAM-1 on ECs, while 15 and 35–40 µm diameter venules exhibit the lowest constitutive expression. This venule size-dependent distribution of ICAM-1 is consistent with functional evidence demonstrating that 25- to 30-µm diameter venules sustain the most intense leukocyte adhesion responses to pro-inflammatory mediators. It remains unclear whether other endothelial CAMs exhibit a similar size-dependent distribution within postcapillary venules [11,33,46,120–123,128–130].
7.1.2. Selectins
Selectins are lectin-like adhesion glycoproteins that mediate leukocyte rolling, which serves to sufficiently reduce the velocity of leukocyte movement along endothelial cell to allow for firm adhesion. P-selectin can be expressed by both EC and platelets, while E-selectin is only expressed by EC. Some vascular beds, such as intestine, exhibit significant constitutive expression of P-selectin, while only skin microvessels exhibit basal expression of E-selectin. P-selectin is normally stored as a preformed pool in EC granules (Weibel–Palade bodies), from which it can be rapidly mobilized to the cell surface by histamine, ROS, and leukotrienes. A slower, more prolonged (transcription-dependent) expression of P-selectin can be demonstrated within 4 hours after exposure to cytokines such as TNF-α. E-selectin, which does not exist in a preformed pool, is entirely under transcriptional regulation and requires up to 3 hours to achieve peak expression. The kinetics and magnitude of expression of P- and E-selectin varies between tissues, with the largest increments in both P- and E-selectin expression noted in the lung, small intestine, and heart after endotoxin challenge, while the brain and skeletal muscle exhibit the smallest responses. Histamine-induced P-selectin expression in all vascular beds is inhibited by a histamine-H1 (but not an H2)-receptor antagonist, indicating that histamine engagement of H1-receptors on ECs results in the mobilization of preformed P-selectin to the endothelial cell surface. The time-related changes in endothelial P-selectin expression after histamine treatment, as well as the temporal responses of P- and E-selectin to endotoxin, are shown for the intestinal microvasculature in Figure 7.2 [120,121,131–140].

FIGURE 7.2
Kinetics of expression of endothelial cell adhesion molecules that mediate either the (A) rolling or (B) firm adhesion/emigration of leukocytes in postcapillary venules of mouse intestine. The rapid, short-lived expression of P-selectin in (A) was induced (more...)
Leukocytes also express an adhesion molecule belonging to the selectin family (L-selectin) as well as counter-receptors for EC selectins (PSGL-1). L-selectin is constitutively expressed on most leukocytes, where it is situated on the tips of microvillus cell surface protrusions at a high density. L-selectin mediates leukocyte rolling by interacting with P- and E-selectins expressed on EC. Upon activation of the leukocyte, L-selectin is rapidly shed from the cell surface via a protease-dependent mechanism. P-selectin glycoprotein ligand-1 (PSGL-1) is the most important selectin ligand expressed on leukocytes. PSGL-1 expressed on neutrophils and monocytes is constitutively active and can bind to P- and E-selectins, as well as L-selectin. On lymphocytes, PSGL-1 must be enzymatically glycosylated for selectin binding. Recent evidence indicates that EC can also express low levels of PSGL-1 [120,121,140–143].
7.1.3. Endothelial Cells Immunoglobulin-Like Adhesion Molecules
ICAM-1, ICAM-2, VCAM-1, and PECAM-1 all belong to a family of immunoglobulin-like molecules that are expressed on the surface of EC. These molecules engage with leukocyte counter-receptors to mediate firm adhesion and/or transendothelial migration. ICAM-1 and ICAM-2 exhibit significant constitutive expression on EC in most vascular beds. When normalized for interorgan differences in EC surface area, the lung exhibits the highest density of ICAM-1, followed closely by the small intestine. Even though other organs (e.g., brain) express much lower levels of ICAM-1, the density of ICAM-1 in all organs is 100 to 1000 times greater than that observed for P- or E-selectin in the same tissues. The observation that constitutive ICAM-1 expression is significantly reduced in microvessels of the GI tract and skin of germ-free mice compared to their conventional counterparts suggests that indigenous gastrointestinal microflora are responsible for a significant proportion of the basal ICAM-1 expression that is detected in both intestinal and extra-intestinal tissues [120,121,140,144–146].
The constitutive expression of ICAM-2 and PECAM-1 on EC in most vascular beds is high, and these adhesion glycoproteins are not up-regulated in response to cytokine challenge. ICAM-1 and VCAM-1, on the other hand, respond to cytokine or endotoxin challenge with a time- and dose-dependent increase in EC expression that is transcription-dependent. The oxidant-sensitive transcription factors, NFkB and AP-1, play a major role in linking EC activation with a variety of different stimuli to increased ICAM-1 and VCAM-1 expression in inflamed microvessels. The transient surge in circulating soluble ICAM-1 (sICAM-1) concentration that precedes the increased surface expression of ICAM-1 on EC suggests that there is a rapid, massive shedding of membrane-bound ICAM-1 from EC throughout the inflamed microvasculature and the eventual clearance of shed protein from the circulation. The shedding of membrane-bound adhesion glycoprotein from activated EC accounts for the use of soluble circulating adhesion molecules as a surrogate marker for EC activation and the severity of inflammatory response [120,121,145,147,148].
7.1.4. Leukocyte Integrins
Integrins are glycoprotein complexes consisting of α- and β-subunits. Once integrin function is activated, the glycoprotein can mediate a strong adhesive interaction (firma adhesion) with counter-receptors on EC. For example, VLA-4 is an α4/β1 integrin that enables monocytes and lymphocytes to bind to VCAM on EC. The β2 (CD11/CD18) integrins play a more important role in mediating the firm adhesion of neutrophils. The CD11/CD18 complex is composed of three structurally and functionally related glycoprotein heterodimers comprised of a distinct α-subunit (CD11a, CD11b, CD11c) that is noncovalently bound to a common β-subunit (CD18). Both CD11a and CD11b are constitutively expressed on the surface of most leukocytes. Most of the CD11b/CD18 and CD11c/CD18 glycoproteins are stored in granules and can be rapidly (within minutes) mobilized to the surface of leukocytes. Following leukocyte activation, some heterodimers (e.g., CD11b/CD18) are mobilized to the cell surface while the constitutively expressed CD11a/CD18 (which is not stored in granules) heterodimer rapidly achieves a high-avidity (active) state due to conformational changes in the glycoprotein. These changes in the amount and avidity of CD11/CD18 on the leukocyte surface allows for a strong interaction of the adhesion molecule with its EC counter-receptor (e.g., CD11a/CD18 to ICAM-1 or ICAM-2). The simultaneous rapid up-regulation of CD11b/CD18 and shedding of L-selectin on leukocytes upon activation (Figure 7.3) enables leukocytes to rapidly transition between the rolling and firmly adherent states [120,121,123,140,149].
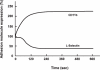
FIGURE 7.3
Time-course of expression of CD11b/CD18 and L-selectin on rabbit neutrophils following exposure to platelet activating factor. Within seconds after PAF exposure, the neutrophils shed L-selectin while rapidly increasing the expression of CD11b. Modified (more...)
The pathophysiological importance of selectins, immunoglobulin-like adhesion molecules, and integrins in mediating the proposed adhesive function in intact postcapillary venules is supported by a large number of animal studies that employed adhesion molecule-specific blocking monoclonal antibodies (mAbs) and/or adhesion molecule-deficient mice to show an attenuated adhesion response. In many of these same studies, evidence is also provided to demonstrate the contribution of leukocyte–endothelial cell adhesion to the microvascular dysfunction (e.g., EC barrier dysfunction) and tissue injury that accompanies acute and chronic inflammation [86,127,140,150,151].
7.2. Chemical Mediators
7.2.1. Pro-Adhesive Mediators
A variety of chemical mediators are released from inflamed tissue that can act on receptors expressed on leukocytes and/or ECs to promote or inhibit leukocyte–endothelial cell adhesion. Histamine, leukotrienes, and platelet activating factor (PAF) production/release from degranulated mast cells and/or activated macrophages lying in proximity to the vessel wall are likely signals for the rapid induction of leukocyte rolling, with histamine inducing the rapid mobilization of preformed P-selectin the EC surface. The leukotrienes (e.g., LTB4) and PAF can also activate rolling leukocytes, increase the expression/avidity of CD11/CD18 and initiate the transition to firm adhesion. Cytokines and chemokines released from perivascular cells and the vessel wall elicit the transcription-dependent expression of endothelial selectin (E- and P-selectin), as well as increasing the expression of ICAM-1 and VCAM-1, which require several hours to achieve peak expression on EC. Activated EC characteristically produce ROS at an accelerated rate, which contributes to transcription-dependent adhesion molecule synthesis/expression by activating key oxidant-sensitive transcription factors (NFkB, AP-1). The changes in EC adhesion molecule expression induced by cytokines and oxidative stress allow for a sustained increase in leukocyte rolling, firm adhesion, and transendothelial migration [120,121].
7.2.2. Anti-Adhesive Mediators
There are several naturally occurring biological agents that appear to serve as endogenous anti-adhesion molecules. These include NO, prostacyclin, and adenosine. Several lines of evidence also implicate NO as an endogenous inhibitor of leukocyte adhesion in venules: (1) NO synthase inhibitors elicit the recruitment of adherent leukocytes, (2) NO donors (nitroprusside, SIN-I) attenuate or prevent the leukocyte adherence induced by different inflammatory stimuli, (3) superoxide, which reacts with NO to render it biologically inactive, promotes leukocyte adherence, and (4) SOD, which scavenges superoxide and limits the inactivation of NO, inhibits leukocyte adhesion. Direct exposure of isolated neutrophils to NO synthase inhibitors does not induce increased expression/avidity of CD11/CD18 nor do they increase the adhesion of leukocytes to biologically inert surfaces (plastic), indicating that these agents promote leukocyte adherence through an action on the endothelium or other cell types. Overall, the available data suggest that any condition that tips the balance between NO production and superoxide generation in favor of the latter will elicit recruitment of adherent leukocytes within postcapillary venules [152–156].
Adenosine and prostacyclin are also very effective in reducing the adherence and emigration of leukocytes in inflamed in postcapillary venules. The anti-adhesive actions of adenosine are mediated through the engagement of A2, but not A1, receptors on leukocytes. Although adenosine-mediated inhibition of neutrophilic superoxide production is also mediated through the A2, receptor, this does not appear to provide the basis for adenosine's anti-adhesive action because SOD is much less effective than adenosine in reducing PAF-induced leukocyte adherence in venules. Adenosine A2-receptor-mediated effects appear to explain the potent reduction in inflammatory mediator-induced leukocyte adhesion and emigration following treatment with methotrexate, which is commonly used in the treatment of rheumatoid arthritis. Prostacyclin (PGI2), which is known for its ability to inhibit platelet aggregation, also appears to act as an inhibitor of leukocyte–endothelial cell adhesion. The view that PGI2 affects leukocyte adherence is supported by the observation that prostaglandin synthesis inhibitors (e.g., indomethacin) promote leukocyte adherence in mesenteric venules, an effect that can be reversed with exogenous PGI. Iloprost, a stable prostacyclin analogue, also exerts a profound inhibitory influence on leukocyte adherence in postcapillary venules exposed to I/R [120,157–163].
7.3. Role Of Hydrodynamic Forces
Shear forces generated by the movement of blood within the microvasculature are generally higher in arterioles than in downstream venules. For example, a 30-μm diameter venules in rat mesentery is likely to exhibit a resting shear rate that is nearly half that of an arteriole of comparable diameter. Since wall shear stress or shear rate represents an anti-adhesion force that opposes the pro-adhesive forces generated by leukocyte and EC adhesion molecules, vessels with a low spontaneous shear rate (low blood flow) would be expected to exhibit more leukocyte adhesion than vessels with high shear rates. For this reason, it has been proposed that leukocytes rarely roll and adhere in arterioles because the higher shear forces exceed the adhesive force in these vessels. Based on this proposal, one might predict that reductions in arteriolar shear rate to levels experienced by venules should promote leukocyte adhesion in arterioles. However, this is not the case because when cat mesenteric arterioles and venules of the same size are exposed to the same range of shear rates (100–1250 sec–1), venules exhibit far more leukocyte rolling and adherence than arterioles. Studies using retrograde perfusion of the microcirculation have also provided some insight into the role of shear rate in the arteriolar–venous differences in leukocyte–endothelial cell adhesion. In the mesentery, retrograde perfusion is associated with a reduced flux of rolling leukocytes in venules and increased leukocyte rolling in arterioles. However, more leukocytes still rolled in venules during normograde perfusion than rolled in arterioles during retrograde flow. These observations indicate that hemodynamic differences between arterioles and venules cannot explain the predilection for leukocyte rolling and adherence in venules and that a more probable explanation for the greater adhesive interactions between leukocytes and venular endothelium is that the counter-receptors (ligands) for leukocyte adhesion molecules are more densely concentrated on venular endothelium [120,140,164–169].
Shear forces can, however, play an important role in modulating the leukocyte–endothelial cell adhesion that occurs during inflammation. The prevailing shear rate exerted on the walls of postcapillary venules determines the level of leukocyte rolling and firm adherence and dictates the contact area between leukocytes and the endothelial cell surface. Even in the absence of an inflammatory stimulus, graded reductions in venular shear rate for brief periods (<2 min) elicit progressive recruitment of both rolling and firmly adherent leukocytes. Similarly, it has been noted that the number of adherent leukocytes recruited into venules by an inflammatory stimulus is inversely proportional to the wall shear rate, suggesting that it is easier for leukocytes to create strong adhesive bonds with ECs at low shear rates and that high shear rates may prevent the creation of such bonds. This effect of venular shear rate on the intensity of the leukocyte–endothelial cell adhesion suggests that the changes in blood flow that are associated with the early and sustained phases of inflammation (discuss above) may exert a significant influence on the intensity of the leukocyte recruitment response during inflammation [165–167].
7.4. Cytotoxicity Of Adherent Leukocytes
Neutrophils that are firmly adherent to vascular ECs are also activated, which results in the production and release of ROS, proteases, cationic proteins (e.g., defensins) and a variety of other chemicals that can impair the function or inflict injury to microvessels. Activated neutrophils utilize the plasma membrane-associated enzyme NADPH oxidase to produce superoxide, which subsequently reacts with itself (spontaneous dismutation) to generate hydrogen peroxide. The potent oxidizing and chlorinating agent hypochlorous acid (HOCl) is also produced when hydrogen peroxide and extracellular chloride ions react with myeloperoxidase (MPO), a cationic enzyme released from neutrophil granules (Figure 7.4). MPO binds avidly to the negatively charged endothelial glycocalyx and is subsequently internalized, with a resultant rise in intracellular ROS. Internalized MPO modulates vascular signaling and impairs vasodilatory function by decreasing the bioavailability of NO through HOCl-mediated chlorination of L-arginine and direct inactivation of NO [170–176].

FIGURE 7.4
Neutrophil-mediated mechanisms of microvascular dysfunction/injury. Upon activation, neutrophils generate superoxide (O2 –) from NADPH oxidase, with a resultant dismutation of O2 – to form hydrogen peroxide (H2O2). The activated neutrophils (more...)
Activated neutrophils also secrete a variety of proteases, which have the potential to elicit uncontrolled proteolysis of vascular wall elements (e.g., basement membrane) and of the interstitial matrix. Many of these proteases are secreted in an inactive (latent) form that is dependent on oxidative mechanisms (HOCl) for activation (Figure 7.4). Extracellular fluid is well-endowed with antioxidants and antiproteases, which limit the cytotoxic potential of circulating neutrophils. However, when neutrophils adhere to ECs, a sequestered microenvironment is created (at the leukocyte–endothelial cell interface), which allows neutrophil-derived oxidants and proteases to overwhelm plasma antioxidants and anti-proteases, thereby enabling the neutrophil to exert its full cytotoxic potential at the vessel wall. The accompanying vascular dysfunction can be manifested as a hyperadhesivity of EC to leukocytes and platelets, the formation of microthrombi, increased production of ROS by EC, and diminished endothelial barrier function [174,175].
The major neutrophil-derived proteases include elastase, collagenase, and gelatinase. These enzymes represent potent mechanisms by which neutrophils may degrade the key components of the endothelial cell basement membrane and interstitial matrix. Elastase, like many proteins released from activated neutrophils, is cationic, which facilitates its interaction with the EC surface (glycocalyx). The metalloproteinases (e.g., gelatinase, collagenase) are secreted in a latent, inactive form that requires further processing for activation. HOCl has been shown to oxidatively activate collagenase and gelatinase secreted by human neutrophils. The role for chlorinated oxidants in activating the metalloproteinase is further substantiated by studies that demonstrate that neutrophils from patients with chronic granulomatous disease (neutrophils are unable to generate ROS) are also unable to activate collagenase. It has also been suggested that activation of gelatinase by chlorinated oxidants is required for neutrophils to degrade the type IV collagen of the endothelial basement membrane and to emigrate from the vasculature. The secretion and subsequent oxidant activation of collagenase would then facilitate the degradation of the interstitial collagens (types I, II, and II), thereby allowing migration of neutrophils within the interstitial matrix [151,172,174,175,177].
The fact that a normal inflammatory response does not result in proteolytic degradation of the interstitium suggests that the host has ways to control this potentially injurious process. Indeed, plasma and interstitial fluid (lymph) contain high concentrations of protease inhibitors. One of best-characterized endogenous protease inhibitors is α1-proteinase inhibitor (also known as α1-antitrypsin). This protein is especially active at inhibiting elastase by forming a complex with the protease and rendering it catalytically inactive. Extracellular fluid also contains other proteinase inhibitors such as α2-macroglobulin and secretory leukoproteinase inhibitor. All of these antiproteases are susceptible to oxidative inactivation by neutrophil-derived oxidants like HOCl. This inactivation is presumed to occur in the subjacent space created by adherence of the neutrophil to cellular membranes or the extracellular matrix. Support for this mechanism is provided by the observation that when the neutrophils are prevented from generating HOCl, the antiproteases remain active and are able to inhibit tissue injury [172–175,178].
The endothelial surface layer (ESL or glycocalyx) appears to be a vulnerable target for the products of neutrophil activation. ROS, MPO, and proteases released by activated adherent neutrophils have the capacity to degrade or depolymerize the membrane-bound glycoproteins and proteoglycans that comprise the glycocalyx. This effectively reduces the thickness of the ESL and creates an opportunity for EC adhesion molecules such as P-selectin to protrude through the ESL and bring the adhesion molecule in close contact with the EC surface. Under normal conditions, the glycosaminoglycan chains and soluble components of the glycocalyx appear to shield adhesion molecules, which prevent the adhesion of circulating cells (leukocyte, platelets) with EC. During inflammation, both EC and adherent neutrophils are activated, resulting in oxidative and enzymatic degradation of the glycocalyx, an opening of the meshwork, and the exposure of EC adhesion molecules, which in turn, amplifies the recruitment of blood cells onto the vessel wall. The leukocyte-mediated ESL degradation also diminishes endothelial barrier function [38–40,179,180].
Chapter 8. Platelet–Vessel Wall Interactions
Platelets are known to adhere and aggregate at sites of vascular injury in response to endothelial denudation and exposure of subendothelial collagen. The accumulation of platelets at the injury site serves to temporarily plug the damaged vessel and localize subsequent procoagulant events. Recent work has revealed, however, that endothelial denudation is not an absolute requirement for platelet attachment to the walls of blood vessels. Although healthy ECs prevent platelet adhesion by hiding components of the subendothelial matrix (collagen, fibronectin) from platelets, augmenting fibrinolysis, and producing platelet inactivators (NO, PGI2), inflammation can lead to an altered phenotype of ECs, leukocytes, and platelets that enhances the capacity of these cells to bind to each other. Consequently, platelet–vessel wall interactions are often observed in the inflamed microvasculature. The recruitment of rolling and adherent platelets in blood microvessels appears to be a well-regulated process that involves the expression and/or activation of adhesion molecules on platelets, ECs, and/or leukocytes. Consequently, platelets can bind to the vessel wall either via a direct interaction with ECs or indirectly by attaching to already adherent leukocytes [12,181–187].
A variety of inflammatory stimuli have been shown to elicit the adhesion of platelets in the microcirculation of different vascular beds, including brain, intestine, mesentery, liver, lung, skeletal muscle, and retina. Some of these stimuli induce the rolling and/or firm adhesion of platelets within a few minutes (calcium ionophore A23187, oxidized LDL), while others require hours (endotoxin, TNF-α), or days (Plasmodium berghei malaria, hypercholesterolemia [HCh]). In most instances, the platelet adhesion response is confined to the postcapillary venules; however, there are several descriptions of platelet adhesion in arterioles, and fewer descriptions of a response in capillaries [12,137,142,180,184,188–190].
8.1. Platelet Activation: Mechanisms And Consequences
Platelet dysfunction is a feature of acute and chronic inflammatory diseases. This is often manifested as an increased expression of activation-dependent surface antigens on circulating platelets, including P-selectin and CD40 ligand. Soluble CD40L levels in plasma are also elevated, which largely reflects the shedding of this pro-inflammatory signaling molecule from the surface of activated platelets. Changes in platelet function during inflammation are also reflected in the increased tendency for platelets to spontaneously aggregate in vitro [20] and to exhibit an increased sensitivity to endogenous pro-aggregation molecules such as collagen and adenosine diphosphate (ADP). The mechanisms that underlie the platelet activation associated with inflammation remain poorly understood [191–194].
It may be expected that as they course through the microvasculature of inflamed tissue, platelets are likely exposed to a variety of substances that either prime the cells for activation or directly activate them. In regions with tissue damage, platelets may be exposed to collagen, a potent stimulant for activation. The direct contact of platelets with cytokine-activated ECs can also lead to platelet activation. Similarly, a variety of soluble substances released from injured resident cells and/or recruited inflammatory cells may also participate in the activation of platelets within the intestinal vasculature. ADP may accumulate in inflamed tissue either as a result of diminished capillary perfusion or due to inhibition of ectonucleotidase CD39, which is expressed on the surface of EC and circulating immune cells, where it efficiently hydrolyzes extracellular ATP and ADP (both of which stimulate platelet adhesion) to AMP and ultimately adenosine (which inhibits platelet aggregation). Oxidative stress and proinflammatory cytokines (e.g., TNF-α) also down-regulate CD39 on T-lymphocytes. Arachidonic acid and PAF produced in response to phospholipase A2 activation are also potential mediators of platelet activation in the inflamed tissue. Once platelet activation is initiated, then a variety of substances that are produced (e.g., thromboxane A2, ADP, serotonin) by platelets, released from granules, or shed (CD40L) from the cell surface can amplify the activation and accumulation of platelets in the microvasculature [187,191,195–197].
An attenuated EC production of endogenous inhibitors of platelet activation, including prostacyclin (PGI2) and NO, may also contribute to the platelet activation response during inflammation. In vitro studies implicate both NO and superoxide as modulators of homotypic platelet aggregation as well as the adhesion of platelets to monolayers of cultured ECs, with superoxide promoting and NO inhibiting the platelet adhesion responses. Platelets and ECs both produce NO from the constitutive isoform of NO synthase (eNOS) and both cell types as well as leukocytes have the capacity to produce superoxide from NADPH oxidase and other enzymes. NO is known to affect the function of different cells via cyclic GMP (cGMP)-dependent and cGMP-independent (related to target specific nitrosation) pathways. Superoxide scavenging may be an important property of NO that enables it to modulate platelet adhesion during inflammation. NO can react with superoxide at a rate that is three times faster than SOD can convert superoxide to peroxide. This avidity of NO for superoxide (and vice versa) indicates that the balance between NO and superoxide fluxes in microvessels may be an important determinant of how either of these reactive species can modulate platelet adhesion during inflammation [184,198–200].
Both NO and superoxide may also indirectly affect platelet adhesion due to their well established effects on leukocyte–endothelial cell adhesion and VSM tone. Superoxide appears to promote, while NO inhibits, leukocyte–endothelial cell adhesion in postcapillary venules. Since leukocyte-dependent mechanisms contribute heavily to the platelet adhesion observed in some pathophysiological states (as discussed below), an imbalance between NO and superoxide may ultimately affect platelet recruitment by modulating leukocyte adhesion. Similarly, because shear forces generated within microvessels can influence platelet adhesion, the ability of NO to produce vasodilation and for superoxide to cause vasoconstriction could account for some of the altered platelet adhesion responses that are observed when the NO-superoxide balance is altered in inflamed tissue.
Activated platelets produce and release a variety of substances that have the potential to influence the quality and intensity of an inflammatory response. These platelet-derived factors act on both leukocytes and ECs to induce an inflammatory phenotype. Some products of platelet activation contribute to transcellular metabolic reactions in neutrophils, which use arachidonic acid released by platelets to produce increased quantities of inflammatory leukotrienes. The attachment of activated platelets to neutrophils also enables the latter produce larger quantities of superoxide and PAF than either cell is capable of producing alone [201–203].
ECs are also an important target for platelets and their activation products. When CD40L-positive platelets are co-incubated with cultured EC, the ECs become activated, as evidenced by an increased surface expression of ICAM-1 and VCAM-1, an enhanced production of IL-8 (a neutrophil chemoattractant), and increased leukocyte–endothelial cell adhesion. These platelets also release the chemokine RANTES, which binds to glycosaminoglycans on the endothelial cell surface to further promote leukocyte adhesion. Since RANTES has been recently implicated as a mediator of impaired endothelium-dependent vasodilation associated with hypertension, it is possible that this platelet-derived chemokine may contribute to the reduced capacity of arterioles in inflamed tissue to dilate in response to acetylcholine and other endothelium-dependent vasodilators [203–207].
8.2. Platelet–Endothelial Adhesion
In some forms of inflammation, the accumulation of platelets within the microvasculature appears to result from a direct interaction between platelets and vascular ECs. For example, endothelial cell activation appears to play an important role in the platelet adhesion response of cerebral and intestinal venules to HCh. Although circulating platelets also assume an activated phenotype in hypercholesterolemic animals and humans, the adhesion of platelets in the microcirculation occurs only when platelets derived from mice placed on a normal (ND) or high-cholesterol (HCD) diet are monitored in HCD-recipient, but not ND-recipient, mice, suggesting that platelet activation is not sufficient to elicit the adhesion response. Using a similar experimental strategy, endothelial cell, rather than platelet, activation has also been invoked to explain the platelet adhesion induced by either bacterial endotoxin (lipopolysaccharide [LPS]) or P. berghei malaria in WT mice or by hypoxia–reoxygenation in sickle cell transgenic mice [189,190,208–210].
Figure 8.1 illustrates some of the possible adhesion receptor–ligand interactions that may account for the adhesion of platelets to microvascular ECs in different pathophysiological states. Platelet–endothelial cell adhesion elicited in acute models of inflammation (e.g., I/R) has been linked to interactions between ICAM-1 on ECs, fibrinogen, and the platelet adhesion molecule GPIIb/ IIIa. In this situation, the oxidative stress experienced by activated EC results in fibrinogen deposition onto constitutively expressed ICAM-1, creating a scaffold on the vessel wall onto which platelets can adhere using GPIIb/IIIa. The inability of platelets derived from patients suffering from Glanzmann's disease, a deficiency in GPIIb/IIIa, to bind to activated EC, as well as the attenuated adhesion response of normal platelets following administration of a fibrinogen or GPIIb/ IIIa blocking antibody support a role for ICAM-1:fibrinogen:GPIIb/IIIa mechanism. Direct adhesion of platelets to EC can also result from interactions between platelet associated GP1bα with either P-selectin or von Willebrand factor (vWF) on ECs. PSGL-1 expressed on platelets can also interact with EC P-selectin to mediate adhesion. Similarly, with the recent discovery that EC can express PSGL-1 (discussed above), platelet-associated P-selectin could also bind to PSGL-1 on EC. The shear force in a vessel is also an important determinant of the adhesion molecules that mediate platelet adhesion in inflamed microvessels. Under low shear stress (<600 sec–1), as is seen in venules, the ICAM-1:fibrinogen: GPIIb/IIIa mechanism is an effective mediator of platelet adhesion, while high shear stress, as experienced in arterioles, favors the engagement of platelet GPIba with vWF [181,211–213].

FIGURE 8.1
Leukocyte-dependent and leukocyte-independent mechanisms of platelet adhesion in inflamed postcapillary venules. Platelets may adhere directly to ECs, using glycoprotein receptors, such as GPIbα and GPIIb/IIIa, that bind to ligands that are expressed (more...)
P-selectin has received considerable attention as a determinant of platelet adhesion in inflamed microvessels. This lectin-like adhesion glycoprotein is normally stored in granular structures of both platelets (α-granules) and ECs (Weibel–Palade bodies), from which P-selectin can be rapidly mobilized to the cell surface upon endothelial cell activation. Some vascular beds (e.g., intestine) exhibit significant basal expression of P-selectin, with little or no basal expression on unactivated circulating platelets. Blocking monoclonal antibodies and P-selectin-deficient mice have been used to implicate P-selectin as a mediator of the platelet adhesion (both rolling and firm adhesion) induced by both acute (e.g., A23187, I/R) and chronic (e.g., HCh, P. berghei malaria) stimuli. The relative contributions of platelet vs. endothelial cell P-selectin to the platelet adhesion response has been addressed in these and other models of inflammation. This has been achieved using either bone marrow chimeras, produced by the transplantation of bone marrow from P-selectin-deficient donor mice into WT recipient mice (or vice versa) or by monitoring the trafficking of P-selectin-deficient platelets in WT recipients (or vice versa). These experimental strategies have revealed that P-selectin expressed on EC appears to be a major determinant of the platelet adhesion in some inflammation models (e.g., sickle cell disease), while both endothelial cell- and platelet-associated P-selectin contribute to the platelet adhesion in others (e.g., HCh, malaria). In more acute models (e.g., I/R), the rapid adhesion response is entirely dependent on endothelial P-selectin, while the slow, time-dependent platelet adhesion response involves both platelet and endothelial cell P-selectin [184,189,190,204,210,212–215].
8.3. Platelet–Leukocyte Adhesion
Platelet–vessel wall interactions in inflamed microvessels can also result from the binding of platelets to leukocytes that are already attached to vascular endothelium. Platelet–leukocyte adhesion can be mediated by different ligand–receptor interactions, including P-selectin (platelet)–PSGL-1 (leukocyte) and GPIbα (platelet)–CD11b/CD18 (leukocytes) interactions (Figure 8.1). Evidence supporting a role for leukocytes in platelet adhesion in inflamed venules is provided by studies demonstrating an attenuation of the adhesion response in animals rendered neutropenic. Similarly, an attenuated platelet adhesion response has been demonstrated in mice that are genetically deficient in either CD18 or ICAM-1 and in WT mice receiving blocking antibodies directed against these adhesion glycoproteins. The data from these studies of leukocyte-dependent platelet adhesion are consistent with a model wherein leukocytes require P-selectin to roll on venular endothelium and subsequently establish firm adhesion via a CD18–ICAM-1 interaction. The adherent leukocytes, which express PSGL-1 and other P-selectin ligands, then create a platform onto which platelets can bind using P-selectin. This model would explain why P-selectin expressed on both platelets and ECs is required for platelet adhesion in some experimental models and why interfering with leukocyte adhesion or rendering mice neutropenic can lead to a concomitant reduction in platelet adhesion in the microcirculation [181,184,186,215–217].
Efforts to simultaneously monitor and quantify platelet and leukocyte adhesion have revealed that a significant proportion of platelets adhering in inflamed venules are attached to adherent leukocytes. The percentage of total adherent platelets that are bound to leukocytes vary among models of inflammation. For example, with I/R or HCh as the inflammatory stimulus, approximately 25% of the platelets bind directly to venular endothelium, while the remaining 75% of the adherent platelets are attached to leukocytes that are bound to the vessel wall. In colonic venules of mice with experimental colitis, nearly 100% of the platelets are bound to adherent leukocytes. The platelets that are directly bound to venular endothelium are unaffected by ablation of either ICAM-1 or CD18 function; however, the accumulation of leukocyte-bound platelets is dramatically reduced following the ablation of these leukocyte adhesion receptors. P-selectin blockade, however, effectively attenuates both the leukocyte-dependent and leukocyte-independent components of platelet recruitment. It is noteworthy that approximately 40% to 50% of the leukocytes that adhere in inflamed venules are platelet-bearing and the remaining 50% to 60% are platelet-free, suggesting that a specific subpopulation of the adherent leukocytes may bind platelets. This possibility is supported by evidence that platelets will avidly bind to neutrophils and monocytes, but not lymphocytes. Alternatively, most of the adherent leukocytes may be neutrophils (as suggested by the studies in neutropenic mice) but roughly half of these adherent neutrophils may achieve an activation state that allows for platelet adhesion. This possibility is supported by evidence implicating neutrophil-derived superoxide in the modulation of platelet adhesion in inflamed venules [184,216–218].
8.4. Platelet–Leukocyte Aggregates
A variety of inflammatory diseases are associated with the appearance of platelet-leukocyte aggregates (PLA) in systemic blood. Although PLA formation is not always correlated with disease activity, this heterotypic cell–cell interaction appears to yield platelets that are more intensely activated than their counterparts that participate in homotypic (platelet–platelet) interactions. The increased expression of P-selectin on activated platelets enables these cells to bind to leukocytes, which constitutively express P-selectin glycoprotein ligand-1 (PSGL-1), the major ligand for platelet P-selectin. The P-selectin-dependent platelet–leukocyte complexes that are observed in inflamed venules may be a precursor of the free-flowing platelet–leukocyte aggregates (PLA) that are detected at increased levels in patients with inflammatory diseases. It has been proposed that PLA are initially formed on the endothelial surface of inflamed microvessels, where they are subsequently dislodged by shear forces generated from the movement of blood. Some of the PLA released into venous blood may not appear in systemic circulation due to entrapment of the aggregates in capillaries of the lung and/or liver. Since leukocyte-free P-selectin-positive platelets also appear in systemic blood, then it is also possible that PLA are formed in flowing blood due to engagement of platelet P-selectin with PSGL-1 that is constitutively expressed on leukocytes. Whether leukocyte activation contributes to the formation of PLA remains unclear. However, platelet activators such as thromboxane and platelet activating factor (PAF) generated by the inflamed tissue may predispose platelets to PLA formation. It has been proposed that the PLA may represent an important circulating source of inflammatory mediators that can sustain or amplify an inflammatory response. Leukocytes with attached platelets appear to be primed for adhesion and can achieve a more activated state than their platelet-free counterparts [187,219–224].
Chapter 9. Coagulation and Thrombosis
Many inflammatory conditions are associated with a hypercoagulable state and a shift in hemostatic mechanisms in favor of thrombosis. While much attention has been devoted to the formation of potentially lethal thromboemboli in large arteries and veins in inflammatory diseases (e.g., atherosclerosis), there is growing evidence from animal models indicating that inflammation also enhances thrombus formation in the microvasculature. Evidence supporting an influence of inflammation on microvascular thrombosis has been derived from experimental models that compare thrombus formation in control and inflamed microvessels that are subjected to injury induced by mechanical trauma, photoactivation, laser light exposure, electrical stimulation, or topical application of caustic chemicals (e.g., ferric chloride). The underlying mechanism of the platelet–vessel wall interactions and thrombus development associated with these models is dependent on whether the injury response is limited to endothelial cell activation (e.g., photoactivation) or involves endothelial denudation and exposure of the subendothelial collagen (e.g., ferric chloride). Nonetheless, both types of thrombosis models have been used to demonstrate an accelerated thrombosis response in arterioles and/or venules during different acute and chronic models of inflammation. Some of the inflammatory stimuli/models studied to date include bacterial LPS, sepsis induced by cecal ligation and puncture (CLP), DSS-induced colitis, HCh, and hypertension [225–232].
9.1. Interdependence Of Coagulation And Inflammation
There is a large and rapidly growing body of evidence suggesting that inflammation and hemostasis are intimately linked processes, wherein each process propagates and intensifies the other, creating the potential for a vicious cycle of thrombogenesis and inflammation (Figure 9.1). The induction of this procoagulant, prothrombotic state likely involves ECs, leukocytes, and platelets, which are activated in response to the inflammatory stimulus. The anticoagulant role of ECs is diminished during inflammation, and this can result from an increased expression of tissue factor (TF, the initiator of coagulation), down-regulation of the anticoagulant protein C pathway, and inactivation of NO by superoxide. The recruitment of rolling and adherent leukocytes on vascular EC can help to create a procoagulant surface for thrombus development. Activated leukocytes also exhibit an increased tissue factor expression and can release proteases that degrade antithrombin as well as cleave and inactivate thrombomodulin on ECs. Tissue factor-rich leukocyte microparticles are also known to contribute to platelet recruitment via P-selectin–PSGL-dependent interactions. The activation and binding of platelets and platelet microparticles to ECs, leukocytes, and to other platelets in the microvasculature of inflamed tissue also promotes a procoagulant state, via an enhanced expression of tissue factor, the generation/activation of coagulation factors (e.g., factor Xa), and enhanced thrombin production [233–238].
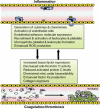
FIGURE 9.1
Vicious cycle of inflammation and coagulation. Inflammation induces a variety of changes in ECs, leukocytes, and platelets, which promote the creation of a procoagulant, prothrombotic surface on the vessel wall (upper). Activation of the coagulation cascade, (more...)
In patients with active IBD, for example, there is evidence for accelerated thrombin generation and increased circulating levels of fibrinogen, vWF, thrombin–antithrombin (TAT) complexes, and clotting factors V, VII, and VIII. In addition, antithrombin III, protein C, protein S, plasminogen activating inhibitor (PAI), and tissue plasminogen activator (tPA) levels are often reduced in these individuals. These manifestations of a hypercoagulable state have also been reproduced in animal models of IBD, such as DSS colitis. In both human and experimental IBD, the hemostatic abnormalities are accompanied by functional changes in circulating platelets that exhibit hyperactivity, hyperaggregability, and a propensity to form platelet–leukocyte aggregates [187,193,194,230,239,240–242].
There is also evidence that the coagulation–anticoagulation pathways exert an influence on the inflammatory response. Different components of the coagulation pathways, including thrombin and tissue factor, appear to promote inflammation, while anticoagulants such as activated protein C (APC) and heparin exert anti-inflammatory effects. Platelets, which are recruited to and activated at sites of thrombus formation, also produce and release a myriad of substances that promote inflammation. The influence of hemostasis on inflammation is supported by numerous reports that describe how different components of the coagulation–anticoagulation pathways can regulate inflammation by exerting an influence on ECs, platelets, and/or leukocytes. Thrombin, for example, has been shown to increase the expression (via transcription-independent and transcription-dependent mechanisms) of adhesion molecules on ECs and to promote leukocyte–endothelial cell adhesion. Also, activation of factor XII can result in complement activation. The engagement of TF with its ligand (factor VIIa) activates the protease-activated receptors PAR1–4, which elicits the production of pro-inflammatory cytokines (TNF-α, IL-6), increases the expression of EC adhesion molecules, and promotes leukocyte rolling in venules. Mice that lack the cytoplasmic domain of TF exhibit an attenuated recruitment of rolling, adherent, and transmigrating leukocytes in postcapillary venules after LPS challenge. Furthermore, a small molecule inhibitor of the TF-VIIa complex (BCX-3607) has been shown to attenuate LPS-induced production of IL-6 and IL-8 in vitro (by ECs) and IL-6 in vivo. Similarly, APC has been shown to inhibit the production of adhesion molecules (VCAM-1, ICAM-1) and cytokines in ECs, as well as agonist-induced leukocyte activation and LPS-induced production of TNF-α and other cytokines by cultured monocytes/macrophages. Finally, mice with single-allele targeted disruption of the protein C gene (heterozygous protein C deficient (PC+/–) mice) have higher levels of circulating cytokines, including a fourfold increase in TNF-α, after endotoxin challenge, which is consistent with an anti-inflammatory action of APC [233,243–246].
9.2. Inflammation-Induced Microvascular Thrombosis: Site-Specific Responses
While both arterioles and venules exhibit the capacity for thrombus formation, some inflammatory stimuli appear to preferentially enhance thrombogenesis in one segment of the microvasculature. For example, venules are far more responsive than arterioles to the thrombosis-enhancing effects of endotoxin (LPS) or sepsis induced by CLP. DSS colitis, angiotensin II-induced hypertension, and HCh, on the other hand, appear to preferentially enhance thrombus formation in arterioles. The basis for this predisposition of arterioles and venules to certain inflammatory stimuli/conditions remains unclear. However, there are a number of characteristic differences in the function and behavior of the two vascular segments that could underlie the differential thrombogenic responses. Thrombi formed in the venous system are characteristically rich in fibrin and trapped red blood cells and poor in platelets, while arterial thrombi are rich in aggregated platelets. Arterioles exhibit a higher shear rate than venules, which would favor the participation of some coagulation factors (e.g., vWF) in the thrombogenic response in arterioles. The higher shear rate in arterioles may also render these vessels vulnerable to inflammation-induced reductions in NO bioavailability. Studies of the concentration profile of platelets within arterioles and venules have revealed that the density of platelets near the vessel wall is much higher in arterioles than in venules, and this difference in platelet distribution cannot be attributed to the occurrence of leukocyte margination in venules [225,228–231,247–249].
Venules differ from arterioles in other ways that could explain the differential thrombogenic responses to inflammatory stimuli. It is likely that the density, distribution and/or production of pro- and anti-coagulation factors (e.g., TF) differs between the vessel types and that venules exhibit less dilution of locally generated procoagulants (e.g., thrombin) by the slower moving blood. The preferential trafficking of leukocytes on venular endothelium during inflammation may render venules more vulnerable to certain inflammatory stimuli. For example, it has been reported that neutrophils may promote thrombosis via the release of neutrophil extracellular traps (NETs) [44]. Bacterial endotoxin as well as activated platelets can induce neutrophils to make NETs in the microvasculature. NETs are abundant in thrombi associated with deep vein thrombosis, and it has been shown that platelets under flow in vitro bind avidly to NETs and are able to promote thrombosis. It has been suggested that the backbone of NETs, which is made of chromatin, provides a structure upon which platelets can adhere, become activated, and aggregate, thereby contributing to thrombus initiation and/or stability. Whether NET formation accounts for the enhanced venular thrombosis induced by LPS remains unclear; however, reports describing no effect of either neutropenia or immunoblockade of adhesion molecules that mediate leukocyte–endothelial cell adhesion in response to LPS on the venular thrombosis response would argue against a role for neutrophil-derived NETs in this model of inflammation-enhanced thrombosis [249–253].
9.3. Chemical Mediators Of Inflammation-Enhanced Thrombosis
While different cell populations (platelets, leukocytes, ECs) and microparticles derived from some of these cells (platelets, leukocytes) have been implicated as factors linking inflammation to thrombosis, a role for chemical mediators that are generated in response to activation of these and other inflammatory cells also appears likely. The involvement of circulating blood cells and/or soluble mediators is supported by reports describing enhanced thrombus formation in tissues distant from the inflammatory site. For example, colonic inflammation in mice results in accelerated thrombosis in arterioles of the cremaster muscle. This may merely reflect the passage of blood cells (e.g., monocytes) and/or microparticles that are activated to produce TF as they course through the gut circulation and eventually transit through extra-intestinal vascular beds, such as skeletal muscle. Alternatively, proteins released (cytokines, chemokines) or shed (e.g., soluble forms of CD40L or the endothelial protein C receptor, EPCR) from EC and/or inflammatory cells within inflamed tissue could mediate the distant (as well as the local) thrombotic response to inflammation [187,241].
Pro-inflammatory cytokines may mediate the enhanced microvascular thrombosis that is associated with inflammation. Several cytokines, including IL-1β, TNF-α, and IL-6, appear to be powerful inducers of coagulation. IL-1β, TNF-α, and IL-6 are known to enhance the expression of tissue factor on ECs and monocytes, down-regulate thrombomodulin, reduce the density of endothelial protein C receptors, and inhibit fibrinolysis on ECs. These cytokine is also known to elicit the shedding of EPCR, thereby producing circulating soluble EPCR that can inhibit protein C activation. TNF-α is also known to increase plasma levels of vWF and to deplete tissue factor pathway inhibitor. Incubation of endothelial cell monolayers with purified recombinant TNF-α elicits a time- and dose-dependent increase in tissue factor procoagulant activity. The cytokine also acts directly on ECs to release both tissue- (tPA) and urokinase-type (uPA) plasminogen activators, while also increasing plasminogen activator inhibitor (PAI-1), with inhibition of fibrinolysis and inadequate removal of fibrin as the net result. Indirectly, TNF-α can lead to impaired anticoagulation by causing leukocytes to release elastase, which could cleave and inactivate thrombomodulin on vascular ECs and antithrombin III. Finally, the ectonucleotidase CD39, which is expressed on ECs, platelets and leukocytes, represents another potential target for the prothrombotic actions of cytokines. TNF-α and oxidative stress are known to down-regulate CD39 expression on leukocytes, which would blunt the efficient hydrolysis of extracellular ATP and ADP, and limit the production of adenosine, which is antithrombotic. Transgenic mice that overexpress CD39 are protected against thrombosis. Consequently, an attenuated expression and/or activity of CD39, which has been described in hypertension, IBD, and HCh, may account for the enhanced thrombus formation in acute and chronic inflammatory states [233–236,254–256–259].
Another member of the TNF superfamily of inflammatory molecules, the CD40/CD40L signaling pathway, may also provide a link between inflammation and coagulation/thrombosis. This pathway has been implicated in the microvascular recruitment of platelets in animal models of IBD and HCh. Upon activation, platelets shed large quantities of CD40 ligand (CD40L), which can promote thrombosis by engaging with its receptor on EC to elicit the increased biosynthesis and expression of EC adhesion molecules and induce tissue factor-dependent procoagulant activity. Soluble CD40L (sCD40L), of which >95% of the circulating level is derived from platelets, also acts as a ligand for the platelet glycoprotein GPIIb/IIIa, and the engagement of CD40L with GPIIb/IIa helps to stabilize the thrombus stabilization and activate more platelets. This contention is supported by reports describing delayed arteriolar thrombosis following vessel injury with FeCl3 in CD40L-deficient mice and the restoration of arteriolar thrombosis when the CD40L–/– mice received recombinant sCD40L. The possibility that CD40L contributes to the hypercoagulable, prothrombotic state in chronic inflammatory diseases is also supported by evidence for increased CD40L expression on circulating platelets and increased soluble CD40L (sCD40L) in plasma of patients with atherosclerosis, rheumatoid arthritis, and IBD. Indeed, sCD40L levels are used as a prognostic marker of thrombotic risk in cardiovascular disease [192,195,196,260,261].
9.4. Reactive Oxygen And Nitrogen Species
Enhanced ROS production and diminished NO bioavailability may also contribute to the enhanced thrombosis that accompanies inflammation. NO inhibits platelet function and prevents thrombosis, while ROS (particularly superoxide) promotes platelet aggregation and thrombosis. A role for ROS in thrombus development has been demonstrated in cerebral arterioles subjected to photoactivation (which generates ROS). In this model, platelet thrombus formation was inhibited by dimethyl sulfoxide (DMSO), a hydroxyl radical scavenger, and by SOD. Thrombus formation induced by iontophoretic application of ADP on mesenteric venules is greatly attenuated by SOD treatment and, to a lesser extent, by catalase administration. The nonselective NOS inhibitor L-NAME, enhanced ADP-induced thrombus growth while L-arginine administration had no effect. NOS inhibition also enhances thrombus development in arterioles and venules injured by photoactivation, while NO donors and NO-independent guanylate cyclase activators (e.g., YC-1) have been reported to dose-dependently inhibit thrombus development in the microvasculature. The enhanced arteriolar thrombosis that accompanies HCh is completely reversed by topical delivery of L-arginine (the substrate for NO production by eNOS). While NO appears to afford protection against thrombus development in both arterioles and venules, it appears that endogenous NO production is more important in inhibiting thrombus development in venules than in arterioles [262–267].
Can I Respond to the Activation of Multi Roll
Source: https://www.ncbi.nlm.nih.gov/books/NBK53380/
0 Response to "Can I Respond to the Activation of Multi Roll"
Post a Comment